A simple comparative analysis with the Fish Tree of Life
Jonathan Chang
Source:vignettes/comparative-analysis.Rmd
comparative-analysis.RmdTo demonstrate the functionality of the fishtree package
and how it integrates well with the rest of the R phylogenetics
ecosystem, this vignette will walk you through a simple comparative
analysis.
Getting and cleaning data
A common hypothesis tested in comparative methods is whether habitat shifts drive rates of diversification in various groups. For example, Santini et al. (2013) tested, among other things, whether reef-associated pufferfishes enjoyed faster rates of speciation compared to their non-reef relatives.
First, load the fishtree package and download the subset
of the Fish Tree of Life corresponding to the taxon of interest. To make
things more interesting we’ll work on the entire order (Tetraodontiformes)
rather than the family in the 2013 study.
library(fishtree)
tree <- fishtree_phylogeny(rank = "Tetraodontiformes")
plot(tree, show.tip.label = FALSE, no.margin = TRUE)
Next, we need to get habitat data and associate it with our
phylogeny. The rfishbase package (Boettiger et
al. 2012) has a variety of convenient functions to access data
recorded by the Fishbase editors. Load the rfishbase
package and retrieve the relevant data in the DemersPelag
field, which identifies whether a species is reef-associated or not,
among other things.
library(rfishbase)
tips <- gsub("_", " ", tree$tip.label, fixed = TRUE)
fb_results <- species(species_list = tips, fields = c("Species", "DemersPelag"))
#> duckdb keeps downloaded extensions and secrets in a temporary directory:
#> ℹ /tmp/Rtmp6rzPqw/duckdb
#> This is removed when the R session ends.
#> • Extensions are re-downloaded each session.
#> • Secrets are lost.
#> ℹ Run duckdb(shared_home = TRUE) (or create ~/.duckdb) to keep them (suitable for most users).
#> ℹ Run duckdb(shared_home = FALSE) to accept the temporary directory (and silence this message).
#> ℹ See ?duckdb_storage for details and alternatives.
#> Joining with `by = join_by(SpecCode)`
fb_results <- fb_results[!is.na(fb_results$DemersPelag), ]
head(fb_results)
#> # A tibble: 6 × 2
#> Species DemersPelag
#> <chr> <chr>
#> 1 Abalistes stellatus demersal
#> 2 Acanthaluteres spilomelanurus demersal
#> 3 Acanthaluteres vittiger demersal
#> 4 Acanthostracion quadricornis reef-associated
#> 5 Acreichthys tomentosus reef-associated
#> 6 Allomycterus pilatus demersalNote that we had to replace the underscores in the tip labels with spaces. This is a common source of errors, so if your analyses don’t seem to work correctly always check whether the functions you’re using expect underscores or spaces.
There’s a lot of data in the DemersPelag field, but we
only want to know if the species is reef-associated or not.
reef <- data.frame(tip = gsub(" ", "_", fb_results$Species),
is_reef = as.numeric(fb_results$DemersPelag == "reef-associated"))
head(reef)
#> tip is_reef
#> 1 Abalistes_stellatus 0
#> 2 Acanthaluteres_spilomelanurus 0
#> 3 Acanthaluteres_vittiger 0
#> 4 Acanthostracion_quadricornis 1
#> 5 Acreichthys_tomentosus 1
#> 6 Allomycterus_pilatus 0We’ve also converted the tip labels back to underscores, since we
need to ensure that the tip labels on our phylogeny match the labels on
our trait data. The geiger package (Pennell et
al. 2014) provides a convenient function that will perform this
check. The name.check function expects row names on our
data object, so we will do that as well.
library(geiger)
#> Loading required package: ape
#> Loading required package: phytools
#> Loading required package: maps
rownames(reef) <- reef$tip
nc <- geiger::name.check(tree, reef)
nc
#> $tree_not_data
#> [1] "Abalistes_stellaris"
#> [2] "Acanthostracion_polygonius"
#> [3] "Chelonodon_patoca"
#> [4] "Chelonodon_pleurospilus"
#> [5] "Chilomycterus_spinosus_spinosus"
#> [6] "Lagocephalus_lagocephalus_lagocephalus"
#> [7] "Meuschenia_scaber"
#> [8] "Monotrete_cochinchinensis"
#> [9] "Monotrete_leiurus"
#> [10] "Ostracion_cubicus"
#> [11] "Ostracion_immaculatus"
#> [12] "Ostracion_solorensis"
#> [13] "Paramonacanthus_filicauda"
#> [14] "Rhinesomus_triqueter"
#> [15] "Sphoeroides_cheesemanii"
#> [16] "Stephanolepis_auratus"
#> [17] "Stephanolepis_hispidus"
#> [18] "Takifugu_fasciatus"
#> [19] "Tetraodon_abei"
#> [20] "Tetraodon_baileyi"
#> [21] "Tetraodon_biocellatus"
#> [22] "Tetraodon_cambodgiensis"
#> [23] "Tetraodon_cutcutia"
#> [24] "Tetraodon_erythrotaenia"
#> [25] "Tetraodon_fluviatilis"
#> [26] "Tetraodon_nigroviridis"
#> [27] "Tetraodon_palembangensis"
#> [28] "Tetraodon_suvattii"
#> [29] "Tetraodon_turgidus"
#> [30] "Tetrosomus_fornasini"
#>
#> $data_not_tree
#> character(0)We’ve identified a mismatch between the tree and the data. We’ll
exclude the tips lacking trait data using drop.tip:
If we also had data that was not in the tree, we could exclude that using the following command, but it isn’t necessary in this case:
Confirm that we have the same number of observations in the tree and the data:
Plotting diversification rates
There are several other data sources available in the
fishtree package, including speciation rates computed via
the DR method (Jetz et
al. 2012). Retrieve speciation rate data:
rates <- fishtree_tip_rates(rank = "Tetraodontiformes")
head(rates)
#> species lambda.tv mu.tv lambda.tc mu.tc
#> 2 Abalistes stellaris 0.08859469 0.01282721 0.09181900 0.02239813
#> 3 Abalistes stellatus 0.08859469 0.01282721 0.09181900 0.02239813
#> 34 Acanthaluteres spilomelanurus 0.10180976 0.01614045 0.16054060 0.06301037
#> 35 Acanthaluteres vittiger 0.10180976 0.01614045 0.16054060 0.06301037
#> 131 Acanthostracion polygonius 0.08549937 0.01178475 0.07697343 0.01359198
#> 132 Acanthostracion quadricornis 0.08549937 0.01178475 0.07697343 0.01359198
#> dr
#> 2 0.11678851
#> 3 0.11909205
#> 34 0.28175984
#> 35 0.28406568
#> 131 0.07606301
#> 132 0.07213440We’re interested in just the dr column, so extract that
and convert spaces to underscores again. Then merge the habitat data
with the speciation rate data.
rates <- data.frame(tip = gsub(" ", "_", rates$species), dr = rates$dr)
rownames(rates) <- rates$tip
merged <- merge(reef, rates)As a quick check our data, let’s plot histograms of the DR rate of reef and non-reef species:
breaks <- seq(min(merged$dr), max(merged$dr), length.out = 30)
hist(subset(merged, is_reef == 1)$dr, col = "orange", density = 20, angle = 135,
breaks = breaks)
hist(subset(merged, is_reef == 0)$dr, col = "purple", density = 20, angle = 45,
breaks = breaks, add = TRUE)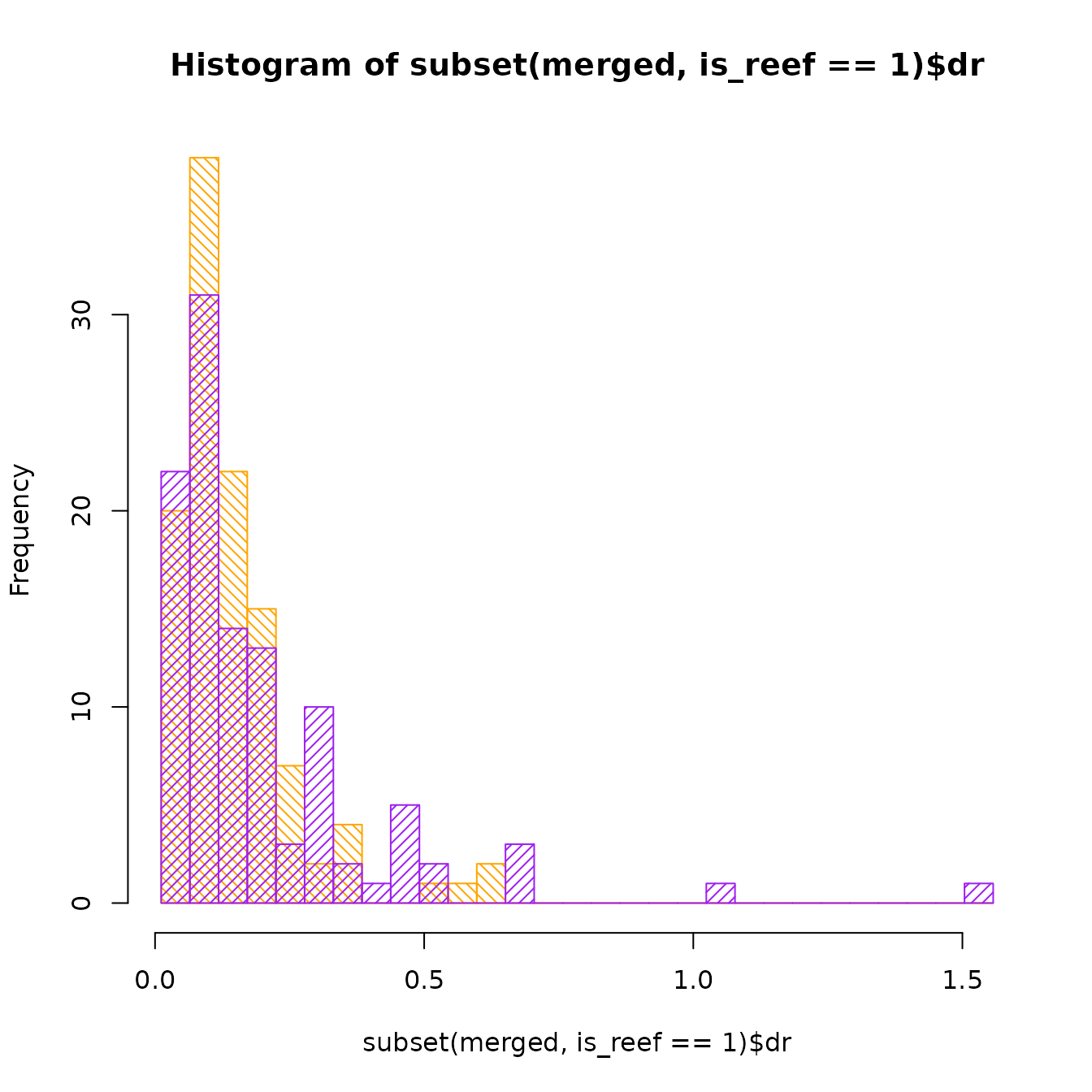
It seems like for the most part, the Tetraodontiformes have a low speciation rate, except for a subset of non-reef species that have a faster rate.
Of course, with any comparative method, it’s critical to consider the historical relationships between the species you’re examining. The following snippet of code is quite complex, but demonstrates how to draw rates onto a phylogeny using colored bars next to each tip in question.
# Plot tree and extract plotting data
plot(tree, show.tip.label = FALSE, no.margin = TRUE)
obj <- get("last_plot.phylo", .PlotPhyloEnv)
# Generate a color ramp
ramp <- grDevices::colorRamp(c("black", "red"), bias = 10)
tiporder <- match(rates$tip, tree$tip.label)
scaled_rates <- rates$dr / max(rates$dr, na.rm = TRUE)
tipcols <- apply(ramp(scaled_rates), 1, function(x) do.call(rgb, as.list(x / 255)))
# Place colored bars
for (ii in 1:length(tiporder)) {
tip <- tiporder[ii]
lines(x = c(obj$xx[tip] + 0.5, obj$xx[tip] * 1.5 + 0.5 + scaled_rates[ii]),
y = rep(obj$yy[tip], 2),
col = tipcols[ii])
}
Running an analysis
Let’s perform a more quantitative analysis using using
hisse. We’ll test 4 models: a BiSSE-like model, a
BiSSE-like null model, a hisse model, and the hisse 2 state null
model.
library(hisse)
#> Loading required package: deSolve
#> Loading required package: GenSA
#> Loading required package: subplex
#> Loading required package: nloptrThe hisse package parameterizes things differently from
diversitree (where BiSSE lives), so we aren’t able to
exactly replicate the analyses in the Santini paper. Instead we’ll
settle by ensuring that the epsilon parameter,
is constrained to be equal for both reef and non-reef taxa. We’ll also
constrain transition rates to be equal, since it can be difficult to
estimate those.
Note that to ensure this vignette can be run in a reasonable amount
of time, we set sann = FALSE to disable the simulated
annealing procedure in hisse. However, for any actual
analysis this option should be turned on for maximum accuracy and
confidence in your final results.
First, we’ll construct and run the BiSSE model and the BiSSE null model:
trans.rates.bisse <- TransMatMakerHiSSE()
pp.bisse.full <- hisse(tree, reef,
hidden.states = FALSE, sann = FALSE,
turnover = c(1, 2), eps = c(1, 1),
trans.rate = trans.rates.bisse)
#> Warning in hisse(tree, reef, hidden.states = FALSE, sann = FALSE, turnover =
#> c(1, : You have chosen to rely on the internal starting points that generally
#> work but does not guarantee finding the MLE.
#> Initializing...
#> Finished. Beginning bounded subplex routine...
#> Finished. Summarizing results...
pp.bisse.null <- hisse(tree, reef,
hidden.states = FALSE, sann = FALSE,
turnover = c(1, 1), eps = c(1, 1),
trans.rate = trans.rates.bisse)
#> Warning in hisse(tree, reef, hidden.states = FALSE, sann = FALSE, turnover =
#> c(1, : You have chosen to rely on the internal starting points that generally
#> work but does not guarantee finding the MLE.
#> Initializing...
#> Finished. Beginning bounded subplex routine...
#> Finished. Summarizing results...Next, we’ll run the full hisse model, save for the constrained transition rates and epsilon.
trans.rates.hisse <- TransMatMakerHiSSE(hidden.traits = 1)
trans.rates.hisse <- ParEqual(trans.rates.hisse, c(1, 2, 1, 3, 1, 4, 1, 5))
pp.hisse.full <- hisse(tree, reef,
hidden.states = TRUE, sann = FALSE,
turnover = c(1, 2, 3, 4), eps = c(1, 1, 1, 1),
trans.rate = trans.rates.hisse)
#> Warning in hisse(tree, reef, hidden.states = TRUE, sann = FALSE, turnover =
#> c(1, : You have chosen to rely on the internal starting points that generally
#> work but does not guarantee finding the MLE.
#> Initializing...
#> Finished. Beginning bounded subplex routine...
#> Finished. Summarizing results...Finally, we’ll build the 2 state character independent diversification model, sometimes called CID-2. We’ll use this as our null model by forcing the visible states (reef or non-reef) to have the same net turnover rates, while permitting the hidden states to vary freely.
pp.hisse.null2 <- hisse(tree, reef,
hidden.states = TRUE, sann = FALSE,
turnover = c(1, 1, 2, 2), eps = c(1, 1, 1, 1),
trans.rate = trans.rates.hisse)
#> Warning in hisse(tree, reef, hidden.states = TRUE, sann = FALSE, turnover =
#> c(1, : You have chosen to rely on the internal starting points that generally
#> work but does not guarantee finding the MLE.
#> Initializing...
#> Finished. Beginning bounded subplex routine...
#> Finished. Summarizing results...We can combine all of our results into a single table for easy comparison.
results <- list(pp.bisse.full, pp.bisse.null, pp.hisse.null2, pp.hisse.full)
aicc <- sapply(results, `[[`, "AICc")
lnl <- sapply(results, `[[`, "loglik")
data.frame(model = c("bisse_full", "bisse_null", "hisse_cid2", "hisse_full"), aicc, lnl)
#> model aicc lnl
#> 1 bisse_full 1823.327 -906.5225
#> 2 bisse_null 1821.332 -906.5726
#> 3 hisse_cid2 1810.644 -901.2283
#> 4 hisse_full 1813.123 -900.3632Summarizing the results on the basis of AICc suggests that the best supported model is a null model, where habitat has no effect on speciation rate.